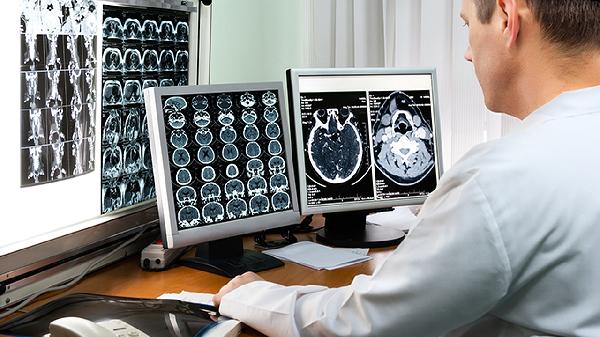
先天性脊柱裂是一种胚胎期神经管闭合不全导致的先天畸形,主要表现为椎管和脊髓发育异常。该病可能由遗传因素、叶酸缺乏、孕期感染、药物暴露、环境毒素等多种原因引起,可通过产前筛查、影像学检查确诊,治疗方式包括手术修复、康复训练及并发症管理。

丙戊酸钠
生产厂家:湖南省湘中制药有限公司
功能主治:本药为原发性大发作和失神小发作的首选药,对部分性发作(简单部分性和复杂部分性及部分性发作继发大发作)疗效不佳。对婴儿良性肌阵挛癫痫、婴儿痉挛有一定疗效,对肌阵挛性失神发作需加用乙琥胺或其他抗癫痫药才有效。对难治性癫痫可以试用。本药除用于抗癫痫外,还可用于治疗热性惊厥、运动障碍、舞蹈症、卟啉症、精神分裂症、带状疱疹引发的疼痛、肾上腺功能紊乱,以及预防酒精戒断综合征。
用法用量:1.成人常用量:每日按体重15mg/kg或每日600~1200mg分次2~3次服。开始时按5~10 mg/kg,一周后递增,至能控制发作为止。当每日用量超过2500mg时应分次服用,以减少胃肠刺激。每日最大量为按体重不超过30 mg/kg、或每日1.8~2.4g。2.小儿常用量:按体重计与成人相同,也可每日20~30mg/kg,分2~3次服用或每日15 mg/kg,按需每隔一周增加5~10 mg/kg,至有效或不能耐受为止。
立即购买
遗传因素是重要诱因之一,部分患者存在基因突变或家族遗传史。孕期补充叶酸可降低风险,但基因缺陷仍需遗传咨询干预。治疗上需针对神经功能缺损进行个性化康复计划,如膀胱功能训练、下肢肌力锻炼。
叶酸缺乏直接影响神经管闭合,孕前3个月至孕早期摄入不足显著增加发病风险。建议育龄女性每日补充400微克叶酸。对于已发病患儿,需评估缺损程度,轻度病例可能仅需定期随访,严重者需手术解除脊髓压迫。
孕期感染如风疹病毒、巨细胞病毒感染可能干扰胚胎发育。孕早期发热超过38.5℃也会增加风险。预防重在疫苗接种和感染控制,出生后患儿若出现脑积水需进行脑室腹腔分流术。
致畸药物暴露涉及抗癫痫药如丙戊酸钠、维A酸类药物等。这类药物可能干扰细胞分化过程,孕期用药需严格评估风险收益比。患儿合并脊柱侧弯时,可能需矫形支具或脊柱融合术。
环境毒素接触包括重金属、有机溶剂等污染物。孕早期接触这些物质可能造成氧化应激损伤。部分患儿伴随脊髓栓系综合征,表现为进行性下肢无力,需通过显微外科手术松解粘连。

约15%患者成年后出现神经源性膀胱,需间歇导尿配合M受体阻滞剂如托特罗定治疗。少数合并Chiari畸形的病例可能需后颅窝减压术。建议所有患者定期进行泌尿系统超声和脊柱MRI随访。